Sequencing data를 얻었을 때 해당 데이터의 quality가 얼마나 좋은지 확인을 해볼 필요가 있다. 이때 가장 일반적으로 사용되는 프로그램 중 하나가 FastQC이다.
1. FastQC 설치
먼저 아래 페이지에 들어가서 FastQC v0.11.9 (Win/Linux zip file)를 다운로드 받고, 압축을 풀어준 이후, 프로그램 폴더를 PATH에 등록해준다 (Java가 없다면 설치를 해줘야 한다 [1]).
https://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc
Babraham Bioinformatics - Public Projects Download
Download Babraham Bioinformatics Projects All Babraham Bioinformatics code is released under the GNU General public license. You should be aware that some of the downloads on this page include code from other projects which is available under different lic
www.bioinformatics.babraham.ac.uk

혹은 아래 명령어를 통해 진행할 수 있다.
$ # sudo apt install default-jre # Java 설치 (Ubuntu)
$ # sudo yum install java-1.8.0-openjdk # Java 설치 (CentOS)
$ wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.9.zip
$ unzip fastqc_v0.11.9.zip
$ PATH=$PATH:/home/FastQC
$ fastqc -help # /home/FastQC/fastqc를 실행한다.
$ which fastqc
/home/FastQC/fastqc
2. FastQC 사용
먼저 예제 데이터를 NCBI SRA에서 다운받는다. 아래 명령어를 통해서 amplicon sequencing data를 다운 받았다. 더 자세한 내용은 아래 글을 참고.
$ fastq-dump -A SRR23192865 --split-3 --gzip
Read 27289 spots for SRR23192865
Written 27289 spots for SRR23192865
$ ls
SRR23192865_1.fastq.gz SRR23192865_2.fastq.gz2021.08.25 - [Bioinformatics/Metagenomics] - [NCBI SRA] 마이크로바이옴 데이터 다운로드 | fastq-dump 설치 및 실행
[NCBI SRA] 마이크로바이옴 데이터 다운로드 | fastq-dump 설치 및 실행
NCBI Sequence Read Archive (SRA)는 마이크로바이옴 데이터(raw sequencing data 및 alignment information)가 저장된 데이터베이스이다. 웹페이지에서 Experiment Accession을 검색하면 해당 실험에 포함된 run file을 다운
bioinfoblog.tistory.com
FastQC의 사용법은 굉장히 간단하다. $ fastqc [file]을 하면 작업 경로에 리포트 파일이 생성된다.
$ fastqc *.fastq.gz
Started analysis of SRR23192865_1.fastq.gz
Approx 5% complete for SRR23192865_1.fastq.gz
Approx 10% complete for SRR23192865_1.fastq.gz
...
Approx 95% complete for SRR23192865_1.fastq.gz
Analysis complete for SRR23192865_1.fastq.gz
Started analysis of SRR23192865_2.fastq.gz
Approx 5% complete for SRR23192865_2.fastq.gz
Approx 10% complete for SRR23192865_2.fastq.gz
...
Approx 95% complete for SRR23192865_2.fastq.gz
Analysis complete for SRR23192865_2.fastq.gz
$ ls
SRR23192865_1_fastqc.html
SRR23192865_1_fastqc.zip
SRR23192865_1.fastq.gz
SRR23192865_2_fastqc.html
SRR23192865_2_fastqc.zip
SRR23192865_2.fastq.gz크게 HTML 파일과 ZIP 파일이 생성되는데 HTML 파일이 리포트이다. ZIP 파일도 압축을 해제하면 HTML 포맷의 리포트 파일이 들어있는데 동일한 파일이며, 추가적으로 fastqc_data.txt 등에서 구체적인 값을 확인할 수 있다.

FastQC는 여러 항목에 대해서 quality를 평가해준다. 이때 HTML 리포트의 오른쪽 summary에서 각 항목에서 어떤 결과(PASS or FAIL)를 받았는지 요약하여 확인할 수 있다. FAIL (X)이면 특히 주의를 가지고 확인할 필요가 있다.
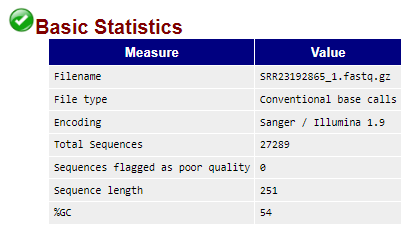
- Basic Statistics: Sequencing read 수, 길이, %GC 등을 확인 가능.
- Per base sequence quality: Read 내에서의 quality 분포를 확인 가능. Quality는 Phred score로 나타내는데, 20점은 99%의 정확도를 의미한다. 일반적으로 20점 이상의 점수는 양호한 품질로 생각할 수 있다.

- Per sequence quality scores: Read의 평균 quality 분포를 확인 가능.

- Per base sequence content: Read 내에서의 ATGC의 분포를 확인 가능.

- Per sequence GC content: Read의 %GC 분포를 확인 가능. 아래는 이론 상의 분포와 실제 분포가 차이가 났기 때문에 결과가 FAIL이 나왔다.

- Per base N content: Sequence 내 N base의 비율

- Sequence Length Distribution: Sequence read의 길이 분포, 해당 파일은 모두 길이가 251 bp였다.

- Sequence Duplication Levels

- Overrepresented sequences: 얼마나 중복된 sequence가 많은지에 대한 값

- Adapter Content

Reference
'Bioinformatics > etc.' 카테고리의 다른 글
| [생물정보학] Sequencing의 종류 (0) | 2021.10.04 |
|---|---|
| [용어 설명] Sequence masking이란? | Soft masking & Hard masking (0) | 2021.10.02 |
| NGS의 구분: Single-end, Paired-end, CCS (0) | 2021.08.12 |
| [용어 설명] SNP (single-nucleotide polymorphism)와 SNV (single-nucleotide variant) 차이 (0) | 2021.08.02 |
| [illumina] Sequence library의 구성 (primer, index, oligo) (0) | 2021.07.30 |