Gene expression Quantification
RNA-seq 등으로 각 샘플에서 각 gene의 read count를 얻었다고 해보자.

이때 1) 샘플 간 read depth가 다르고, 2) gene마다 gene length가 다르기 때문에 이를 normalization할 필요가 있다. Gene expression의 양을 비교할 때 사용하는 normalization 방법에는 RPKM, FPKM, TPM이 있고, TPM이 주로 사용된다.
한편 gene (transcript) length의 경우, 길이가 길수록 fragment도 더 많이 나올 것을 가정하여 normalization을 하지만, 실제로 gene (transcript) length와 fragments의 양이 완전히 비례하지는 않는다. 예를 들어, 길이가 10 bp인 gene으로부터 길이가 5 bp인 fragment는 10개 아니라 10-5+1=6개가 나올 수 있다. 그래서 요즘에는 단순히 gene (transcript) length (위 예제에서 10 bp)를 고려하는 것이 아니라 effective length (actual length - fragment length + 1, 위 예제에서 6)를 normalization에 고려한다고 한다.
RPKM
먼저 RPM (Reads per Million): Sample의 raw read counts를 Scaling factor(Total reads/1M)로 나눈 값이고, RPKM (Reads per kilobase per Million mapped)은 RPM값을 gene length(Kb)로 나눈 값이다.
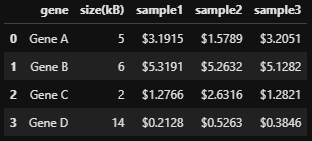
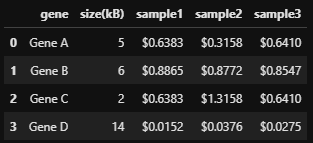

FPKM
FPKM (Fragments Per Kilobase of transcript per Million mapped reads)은 RPKM에서 추가로 paired-end information(Illumina)를 고려하는 것이다.

TPM
TPM (Transcripts per millions): RPKM에서 RPM을 구한 후 gene length로 normalization하는 순서를 바꿔서, 먼저 RPK를 구하고 여기에서 scaling factor를 계산하여 나눠준다.
Sample 간 비교를 할 때, RPKM(or FPKM)은 total read의 개수가 다르게 나오지만, TPM에서는 똑같아서 sample 간 비교가 용이하다.

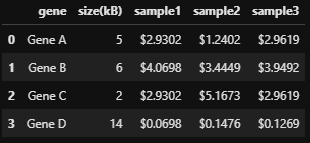
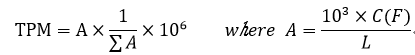
하지만 실제로는 TPM도 샘플 간 비교에 적당하지 않다. 두 샘플의 RNA-seq 데이터가 있을 때 아래처럼 MA plot을 그려볼 수 있다. MA plot은 데이터를 M (log ratio) scale과 A (mean average) scale로 나타내어 그려준다는 의미에서 이름 붙여졌다. 즉, 각 점은 유전자를 나타내고, X축은 두 샘플에서의 발현량 평균(log2 scale)을, Y축은 foldchange (log2 scale)을 의미한다. 일반적으로 두 그룹의 RNA-seq 데이터를 비교할 때 모든 유전자가 다 바뀌었을 것이라고 생각하지는 않는다. 즉, 대부분의 유전자는 Y=0 값을 가지고, 그래프가 대칭일 것으로 기대한다. 하지만 왼쪽의 normalization 전 MA plot을 보면 대부분의 유전자가 -1 쪽으로 치우쳐진 것을 볼 수 있다. 그래서 위 전제를 바탕으로 normalization을 진행할 수 있는데 가장 많이 사용되는 방법이 TMM이며, plot에서의 평균과 Y=0 line을 비교하여 shifting을 해준다. 오른쪽의 normalization 후 MA plot을 보면 대부분의 유전자가 Y=0에 몰려있는 것을 확인할 수 있다.

Python code for RPKM & TPM
def calculateRPKM(df):
df_rpkm=df.copy()
for i in [2,3,4]:
# step 1 compute RPM
df_rpkm.iloc[:,i]=df_rpkm.iloc[:,i]/(df_rpkm.iloc[:,i].sum()/10)
# step 2 normalize for gene length
df_rpkm.iloc[:,i]=df_rpkm.iloc[:,i]/df_rpkm.iloc[:,1]
df_rpkm.iloc[:,i]=df_rpkm.iloc[:,i].map('{:,.4f}'.format)
del df_rpkm['size(kB)']
print(df_rpkm.to_csv(index=False))
print()
def calculateTPM(df):
df_tpm=df.copy()
for i in [2,3,4]:
# step 1: normalize by gene length
df_tpm.iloc[:,i]=df_tpm.iloc[:,i]/df_tpm.iloc[:,1]
# step 2: normalize by mapped reads (per million)
df_tpm.iloc[:,i]=df_tpm.iloc[:,i]/(df_tpm.iloc[:,i].sum()/10)
df_tpm.iloc[:,i]=df_tpm.iloc[:,i].map('{:,.4f}'.format)
del df_tpm['size(kB)']
print(df_tpm.to_csv(index=False))
print()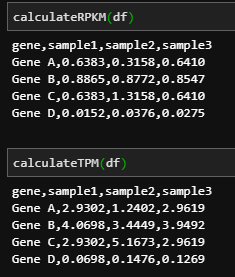
'Bioinformatics > Transcriptomics' 카테고리의 다른 글
| KEGG 데이터베이스에서 유전자 검색 | KEGG 데이터베이스 구조 (0) | 2021.08.17 |
|---|---|
| 특정 유전자 리스트와 관련 있는 pathway 검색 방법 (KEGG Mapper) (0) | 2021.07.05 |
| [Python] Find Open Reading Frame (ORF) | 파이썬 코드 (0) | 2021.06.14 |
| [Prodigal] 원핵생물의 유전자 예측 프로그램 (Prokaryotic Gene prediction) (16) | 2021.05.07 |