Amplicon data는 PCR이나 Sequencing 과정 중에서 발생한 error reads를 포함한다. 우리는 그중에서 correct한 sequence만을 뽑아내서 샘플이 어떻게 구성되어 있는지 파악하기를 원한다. 전통적인 방법으로는 Opertaional taxonomic unit (OTU) picking이 있는데, 최근 들어 Amplicon sequence variant (ASV) pipeline이 각광을 받고 있다. 그러면 OTU는 무엇이고 ASV는 무엇인지, 어떤 차이가 있는지 본 포스팅에서 살펴보고자 한다.
Operational Taxonomic Unit (OTU)
Reads를 97% similarity를 기준으로 묶는 방법(clustering)을 OTU picking이라고 하고, 묶여진 그룹(cluster)를 OTU라고 한다. 이러한 OTU의 목적은 1) taxonomic concepts를 high-throughput marker-gene sequencing에 적용하기 위해서, 2) amplicon sequencing error의 영향을 줄이기 위해서(errors를 error-free sequence와 함께 묶어준다)이다. 비슷한 여러 종이 하나의 OTU로 묶일 수 있고, 각각의 identifications는 잃어버리게 된다 (Abstract of a cluster). OTU picking 방법에는 크게 1) Closed-reference OTU picking, 2) De novo OTU picking, 3) Open-reference OTU picking이 있다.
Closed-reference OTU picking
Closed-reference OTU picking은 sequence reads를 먼저 Reference database에 mapping해보는 방식이다. Reference DB에 존재하는 reads와 97% 이상의 similarity를 갖는 reads만 다루고, 그 외에 reads는 버린다. 이 방식은 DB의 에러나 biases에 취약하다. 또 DB에 없다면 정보를 잃어버리고, diversity measure가 제대로 되지 않는다. 그리고 Often-absent reference sequence가 representative로 역할을 수행할 수도 있다. 그렇지만 샘플 분석에 서로 같은 database가 이용되었다면 비교할 수 있고, 새로운 데이터를 예상하는 biomarker를 찾을 수 있고, 데이터 분석을 병렬적으로 할 수 있으며 계산 자원이 적게 소요된다.
De novo OTU picking
Database 없이 샘플에 존재하는 reads들을 97% similarity 기준으로 묶는 방식이다. 샘플마다 묶이는 방식이 달라지므로 샘플 간이나 study간 비교가 불가능하다는 단점이 있따. 물론 diversity metric과 같은 coarser level 비교는 가능하다. 또 biomarker를 찾을 수 없고, 모든 데이터가 함께 분석되어야 하므로, 계산 자원이 많이 소모된다. 데이터가 추가되거나 제거되면 다시 돌려야 한다. 그렇지만 분석 샘플에 상관 없이 (연구가 활발히 이루어진 host이든 아니든) biological variation을 비교적 정확히 알아낼 수 있다.
Open-reference OTU picking
Closed-reference OTU picking 방식처럼 DB에 먼저 reads를 mapping하고, mapping 되지 않는 reads에 한해서만 De novo OTU picking 방식으로 묶는 방식이다. 즉, 두 방식의 중간 어딘가에 존재하는데, 이는 샘플의 특성에 따라 달라진다. 예를 들어, 연구가 활발히 이루어진 human gut 샘플의 경우에는 closed-reference method와 비슷하게 수행된다.
종합적으로 Closed-reference OTU picking과 De novo OTU picking 방식을 비교하여 정리하면 다음과 같다.
| Closed-reference | De novo | |
| Reproducible | O | X |
| Comprehensive | X | O |
Amplicon Sequence Variant (ASV)
ASV는 high-abundance sequence와 유사한 low-abundance sequence는 error일 것이라는 전제를 바탕으로 구한다. Exact sequences로 결과를 나타내는 것으로서, sequence cluster나 그 cluster를 대표하는 consensus sequence를 다루는 OTU 방법과는 차별점이 있다 (Resolution이 더 높다). Exact sequences는 clustering이나 refence database 없이 생성되기 때문에 reference bias가 없고, 샘플 간 비교가 가능하다. 또한 모든 biological variation을 찾아낼 수 있다. 즉, ASV pipeline은 closed-reference OTU picking과 De novo OTU picking 방식 장점을 모두 가졌다 (Reproducible O, Comprehensive O). Chimera dection에서 OTU picking에서는 97% similarity로 같은 종으로 묶인 reads 중에 chimera가 있더라도 제거할 수 없지만 (이미 같은 OTU로 합해짐), ASV pipeline에서는 제거할 수 있다.
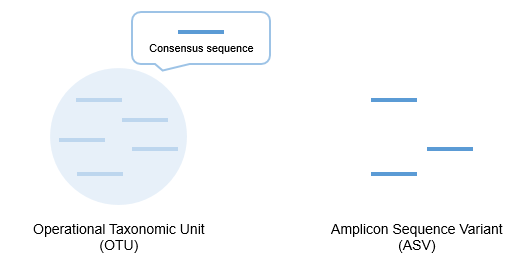
알아두어야 할 점은 ASV pipeline은 각각의 read에 대해 독립적으로 적용할 수는 없고 (Sample 단위에서 적용을 해야한다), 다른 연구 간 비교가 가능하지만, 실제로는 똑같은 primer set를 사용한 경우에만 가능하다. 또한 같은 genome이 multiple ASVs를 가질 수 있고, Short genetic barcode가 가지는 한계를 없애지 못한다 (복잡한 생물을 나타내기에 역부족일 수 있다). 그리고 실제로는 error reads의 count가 높게 나오면, biological sequence라고 판단할 위험이 존재한다 (false positive). 반대로 적게 존재하는 biological sequence를 drop할 수 있다 (false negative).
정리하면,
- ASV pipeline은 OTU picking보다 에러를 더 잘 제거한다.
- ASV pipeline은 OTU picking보다 해상도가 더 높다 (nucleotide level).
즉, 대부분의 논문에서 정확도와 해상력의 측면에서 OTU보다 ASV를 사용하는 것을 추천하는 분위기이다. ASV는 ESV (exact sequence variant), zOTU (zero-radius OTU), SV (sequence variant), actual SV, 등으로도 불린다.
ASV pipeline
ASV pipeline에는 유명한 세 개의 pipeline이 존재한다 (DADA2, Deblur, UNOISE3). 이를 표로 정리하면 다음과 같다.
| Pipeline | Characteristic | Running time | Sample pooling | Implemented in | Open source |
| DADA2 | Sensitive | Fast | O | R | Yes |
| Deblur | Specific | Faster | X | Python | Yes |
| UNOISE3 | Best balance | Fastest | O | C++ | No |
DADA2는 sensitive하고 (rare한 sequence를 더 잘 찾아냄, false positive의 위험이 높음), Deblur는 specific(더 정확하게 sequence를 찾아냄, false negative의 위험이 높음)하다. 어느 프로그램을 선택할지는 연구의 목적에 따라 달라진다.
QIIME DADA2를 사용할 때는 denoising 전 paired-end reads를 join하면 안된다. DADA2는 denosing 과정 중 reads를 join한다. Denoising 전 quality filering 과정도 자동으로 해준다.
QIIME Deblur를 사용할 때는 paired-end reads를 join해야 한다. 또 denoising 전 quality filering 과정을 거쳐야한다.
Reference
- https://www.zymoresearch.com/blogs/blog/microbiome-informatics-otu-vs-asv a
- Callahan, Benjamin J., Paul J. McMurdie, and Susan P. Holmes. "Exact sequence variants should replace operational taxonomic units in marker-gene data analysis." The ISME journal 11.12 (2017): 2639-2643.
- Edgar, Robert C. "UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing." BioRxiv (2016): 081257.
- Nearing, Jacob T., et al. "Denoising the Denoisers: an independent evaluation of microbiome sequence error-correction approaches." PeerJ 6 (2018): e5364.
'Bioinformatics > Metagenomics' 카테고리의 다른 글
| [QIIME2] Moving Pictures 튜토리얼 (QIIME 분석에서 가장 기본이 되는 내용) (0) | 2021.03.02 |
|---|---|
| [QIIME2] 숙련된 연구자들을 위한 QIIME2 Overview (15) | 2021.03.02 |
| [QIIME2] QIIME2의 workflow 설명 (일반적인 Amplicon data 분석 과정) (0) | 2021.02.18 |
| [QIIME2] QIIME2의 핵심 개념 소개 (0) | 2021.02.18 |
| [sORF] 장내미생물이 만드는 Small proteins의 기능 연구 (0) | 2021.02.10 |